Gastrointestinal stromal tumors (GISTs) are a group of mesenchymal malignancies that represent the most common gastrointestinal sarcomas. Interstitial cells of Cajal, which act as pacemakers to regulate intestinal motility, are thought to be the origin of GISTs.
The incidence of GIST is approximately 10-15 cases per million people worldwide, with a median age at diagnosis of 66-69 years and a similar distribution between men and women.
GISTs most commonly arise in the stomach (55.6%), followed by the small intestine (31.8%), colon/rectum (6.0%), other abdominal sites (5.5%) (so-called extragastrointestinal GIST/E-GIST), and the esophagus (0.7%). Approximately 20% of cases are metastatic at the time of diagnosis, with the liver and peritoneal cavity being the most common sites.
Historically characterized as a chemoresistant entity potentially curable only by surgery, GISTs have evolved into a framework for the concept of precision oncology. The characterization of activating mutations in the potentially actionable oncogene KIT in 1998 has paved the way for major diagnostic and therapeutic breakthroughs in this disease, first by using imatinib in 2001, and currently with a growing number of treatment options.
This review will thoroughly discuss the molecular aspects of GISTs and how they translate into clinical practice. Bibliographic searches were conducted on MEDLINE by using PUBMED as the query interface. No language restrictions were applied.
Histologic variants of GIST include spindle cell (60% of cases), epithelioid (30%), and mixed subtypes (10%).
Potential differential diagnoses for GIST include leiomyoma, leiomyosarcoma, schwannoma, peripheral nerve sheath tumor, solitary fibrous tumor, synovial sarcoma, and sarcomatoid carcinoma.
The discovery of oncogenic drivers in GIST has led to a better understanding of the disease and has allowed the development of targeted therapeutic agents. It was not until 1998 that activating mutations in KIT were recognized as a key mechanism in tumor pathogenesis.
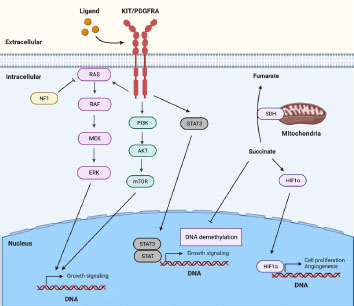
Figure 1. Key signaling pathways involved in GIST pathogenesis. Most GISTs are characterized by gain-of-function mutations in KIT or PDGFRA, resulting in the activation of downstream signaling cascades such as the MAPK, PI3K, and STAT3 pathways. In addition, SDH deficiency contributes to GIST development by activating HIF1a and inhibiting DNA demethylation.
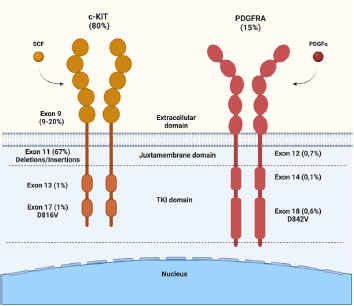
In approximately 10-15% of cases, mutations are found in a specific tyrosine kinase (TK) domain, the platelet-derived growth factor receptor alpha (PDGFRA). These alterations are more common in gastric tumors (particularly in exon 18 D842V) and are generally associated with lower risk of metastasis than KIT-mutated neoplasms, with only 2% of advanced GISTs harboring PGFRA mutations. Taken together, KIT and PDGFRA mutations account for approximately 85-90% of GISTs cases and are considered mutually exclusive.
KIT and PDGF receptors share structural similarities that allow their function to be understood by analogy (
Figure 2. Activating mutations in the KIT and PDGFRA domains.
When mutated, exon 9 (8-10%) in the extracellular binding site leads to ligand-independent receptor dimerization. These mutations are more commonly associated with intestinal tumors. Exons 13 and 17, located in the ATP-binding domain and activation loop, respectively, induce a conformational change to accommodate ATP binding and activate the kinase domain through phosphorylation of its substrates, mediating various pathways such as MAPK and mTOR. When mutated (rare), they allow for disordered ATP binding.
GISTs with PDGFR-driven mutations are more common in the stomach, with a more indolent disease profile and lower rates of distant metastasis compared to KIT-mutant GISTs.
Approximately 10-15% of GISTs that are wild type (wt) for KIT and PDGFRA further divide based on the status of the succinate dehydrogenase (SDH) complex.
SDH deficiency is the most common subtype of KIT and PDGFRA wild-type tumors. Mutations in genes encoding SDH subunits or epigenetic suppression of SDH expression are different mechanisms that can cause SDH deficiency. These SDH-deficient GISTs are more common in children and young adults and generally present with a phenotype of multinodular gastric lesions, lymphovascular invasion, and liver and lymph node metastases. However, despite the conventional risk scheme stratification, they tend to exhibit a more indolent disease profile. Furthermore, this protein deficiency is associated with some genetic predisposition syndromes, most notably Carney-Stratakis syndrome.
Conversely, SDH-proficient GISTs represent a population with various actionable drivers, including mutations in NF1, RAS, BRAF, and PIK3CA genes as well as fusions in NTRK and fibroblast growth factor receptor (FGFR) genes (
| Genetic type | Relative frequency (%) | Anatomical site |
|---|---|---|
| KIT mutation | 80-85 | |
| Exon 9 | 10 | Small intestine, colon |
| Exon 11 | 67 | All sites |
| Exon 13 | 1 | All sites |
| Exon 17 | 1 | All sites |
| PDGFRA mutation | 5-8 | |
| Exon 12 | 1 | All sites |
| Exon 14 | <1 | Stomach |
| Exon 18 D842V | 5 | Stomach, mesentery, omentum |
| Exon 18 other | 1 | All sites |
| Exon 17 | 1 | All sites |
| Wild type | 12-15 | |
| BRAF V600E | 7-15 | Small intestine, stomach |
| SDHA, SDHB, SDHC, SDHB | 2 | Small intestine, stomach |
| Carney triad | Rare | Stomach |
| NF1 | Rare | Small intestine |
| NTRK fusion | Rare | Small intestine, rectum |
Abbreviations: PDGFRA = Platelet-derived growth factor receptor; GIST = Gastrointestinal stromal tumor; NF 1 = Neurofibromatosis type 1; SDH = Succinate dehydrogenase.
Surgical resection remains the cornerstone of the treatment for localized GIST, and complete resection can be achieved in approximately 86% of patients with primary tumors. Unfortunately, at least 40% of these patients will develop relapse within approximately 2 years of follow-up postsurgical treatment, except for small tumors (usually less than 1 cm) that are found incidentally. The main sites of relapse are the liver and peritoneum, which add to the morbidity of the patient. Selection of patients who may benefit from postoperative treatment depends on determining the risk of GIST relapse after curative resection. However, the rarity of this entity and its relatively recent molecular characterization hamper this process.
In 2008, an updated risk stratification system based on the 2001 NIH consensus was published by Joensuu et al., which has also been relevant in refining the selection of patients for adjuvant systemic treatments according to an improved risk stratification. This classification includes tumor size, mitotic activity (in an 5mm2 area, generally equivalent to 50 high-power fields in older microscopes), and primary site (
| Risk category | Size (cm) | Mitotic rate (per 50 HPF) | Primary tumor site |
|---|---|---|---|
| Very low risk | <2 | ≤5 | Any site |
| Low risk | 2.1-5 | ≤5 | Any site |
| Intermediate risk | 2.1-5 | >5 | Stomach |
| 5.1-10 | ≤5 | Stomach | |
| High | Any | Any | Tumor rupture |
| >10 | Any | Any | |
| Any | >10 | Any | |
| >5 | >5 | Any | |
| 2.1-5 | >5 | Nongastric | |
| 5.1-10 | <5 | Nongastric |
Abbreviations: GIST = Gastrointestinal stromal tumor; HPF = High power field.
While the molecular aspects of GISTs are not currently included in the stratification tools, their prognostic value is well established and was demonstrated in a study that analyzed a series of 451 untreated primary localized GISTs for KIT, PDGFRA, and BRAF mutations, revealing that the mutational status is a significant prognostic indicator of overall survival (OS). In this study, KIT-mutated group had a worse outcome than the PDGFRA-mutated and triple negative GISTs (KIT, PDGFRA and BRAF) ones. Therefore, the inclusion of molecular data and risk stratification criteria may help improve decision making, especially in the adjuvant setting.
Endoscopic biopsy may be performed if GIST is suspected. However, diagnostic biopsy is unnecessary when there are clear indications for immediate surgical resection, such as lesions larger than 2-5 cm or exhibiting suspicious radiological findings (e.g., irregular margins, heterogeneous lesions, ulceration, echogenic foci, and cystic degeneration), regardless of size. Furthermore, percutaneous biopsy approaches should be avoided due to the risk of capsule rupture, which is associated with an increased risk of relapse and dissemination.
Typically, esophageal or gastric lesions less than 2 cm are very low risk; in this setting, an active surveillance strategy should be discussed. Although there is no precise optimal schedule for the frequency of this surveillance, most guidelines recommend an initial short-term surveillance within six months and annual surveillance thereafter. Surgical excision can be deferred if the patient becomes symptomatic or if tumor growth is confirmed.
If a resection with microscopic margins cannot be achieved initially, clinicians may consider neoadjuvant therapy with imatinib after multidisciplinary discussion. However, the exact duration of this treatment remains to be determined.
Since approximately 50% of patients who undergo surgery experience disease relapse, the use of imatinib (a specific inhibitor of KIT protein, PDGFRA, and ABL) as an adjuvant treatment has been investigated to improve the prognosis of highrisk patients after curative surgery.
In the double-blind, controlled Z9001 trial, 713 patients with GISTs measuring 3 cm or more within 70 days of total or partial resection were randomized to receive adjuvant imatinib 400 mg daily or placebo for one year. The estimated 1-year relapse-free survival (RFS) rate was 98% vs. 83% for the imatinib and placebo groups, respectively (hazard ratio [HR] = 0.35; 95%CI = 0.22 to 0.53; p<0.0001). However, no improvement in OS was demonstrated, which may be attributed to a favorable response to imatinib after crossover in the control group and possibly suboptimal patient selection, as the inclusion criteria were based solely on tumor size. Features significantly associated with a shorter RFS included a tumor size greater than 10 cm, small intestine location, and high mitotic rate (≥10 per 11.87mm2). In addition, the analysis showed no statistically significant association between RFS and the presence of KIT exon 9 mutation, KIT exon 11 deletion, or PDGFRA mutation.
The multicenter phase III EORTC-62024 trial randomized 908 patients who had undergone total or partial resection of localized GISTs with positive immunostaining for KIT to receive two years of adjuvant therapy with imatinib or observation. The secondary endpoint of RFS rate at five years was 70% in the imatinib group vs. 63% in the observation group and 63% vs. 61% at ten years (HR = 0.71, 95%CI = 0.57 to 0.89, p=0.002), respectively. However, the primary endpoint of 10-year imatinib failure-free survival (IFFS) was 75% vs. 74% and did not reach statistical significance (HR = 0.87, 95%CI = 0.65 to 1.15, p=0.31) regardless of risk according to 2022 NIH classification (tumor size >5 cm and mitotic index >5/50 HPFs). A weakness of this study was the high proportion of patients with intermediate, low, and very low risk (42%), which resulted in numerous protocol and primary endpoint adjustments (change from OS to IFFS) to maintain statistical power in high risk patients, and outcome data based on mutation status were not provided.
The current standard of care for adjuvant treatment of GIST was determined by the Scandinavian Sarcoma Group (SSG) XVIII/AIO phase III trial.
The extension of adjuvant imatinib beyond 3 years was investigated in the single-arm, phase II PERSIST-5 study. It included 91 patients from 21 centers who were treated with imatinib 400 mg once daily for five years or until disease progression or the occurrence of limiting toxicity. The estimated 5-year RFS was 90% (95%CI = 80 to 95%), and the 5-year OS was 95% (95%CI = 86% to 99%). However, only 51% of patients completed five years of imatinib therapy as per the protocol. After molecular analysis, none of the patients with imatinib-sensitive mutations experienced a relapse over five years. In addition to being an uncontrolled, phase II study, other significant limitations include the number of patients with intermediate risk of relapse (26%) and the inclusion of patients with mutations potentially resistant to imatinib, such as PDGFR D842V, RAF, PI3K, NF1 mutants or with SDH deficiency. Therefore, there are still insufficient robust trials to justify standardizing this approach in the adjuvant setting. Randomized, phase III studies with well-defined selection criteria are expected to provide insight into the impact of extended therapy beyond 3 years of duration (NCT 02413736 and NCT02260505).
In contrast to the advanced scenario, in the adjuvant setting, the use of imatinib at 800 mg daily did not result in improved outcomes in a retrospective series of 185 surgically resected GIST patients with exon 9 mutations (
Neoadjuvant or perioperative treatment is not a standard approach for resectable GIST; however, encouraging outcomes that include high complete resection rates, RFS, OS, and higher rates of organpreserving surgery suggest a potential clinical benefit in select scenarios.
Currently, the NCCN reserves the neoadjuvant strategy for patients considered “resectable with significant morbidity”. In this context, molecular evaluation is also necessary to determine the choice of targeted therapy. Several randomized, phase III studies generally support using 400600 mg of imatinib. The appropriate duration of neoadjuvant treatment is still unclear. Regarding advanced/metastatic GIST, the median response time in patients with at least a partial response to imatinib in the B2222 trial was 2.7 months, with 75% of patients requiring 5.3 months to confirm an objective response.
Another point of controversy is the optimal adjuvant treatment in patients undergoing neoadjuvant therapy. Although there is a lack of standardization among studies, there is a tendency to recommend adjuvant treatment in higher-risk patients despite the pathologic response.
The main limitation in neoadjuvant therapy for GIST is the challenge of accurately assessing response to treatment. Imatinib-induced changes in tumor density may not align with traditional size-based criteria such as RECIST. Integrating technologies such as positron emission tomography- computed tomography with fluorodeoxyglucose (FDG PET-CT) has helped to improve the assessment of post-neoadjuvant response to treatment.
Imatinib was approved by the Food and Drug Administration (FDA) in 2002 for use in the advanced scenario based on the B2222 trial, a phase II trial that assessed 147 patients who received either 400 or 600 mg of imatinib. Results showed that approximately 54% of patients experienced a partial response, while 28% had stable disease, and no cases of complete response were observed. There was no difference in results between the two arms in this study. Adverse events reported by participants included edema (74%), nausea (52%), diarrhea (45%), and myalgia (40%). Of note, gastrointestinal or intra-abdominal bleeding occurred in 5% of cases, particularly in patients with a large mass. Nine- year follow-up results from this trial showed similar estimated OS and time to progression.
| First author, study information | Clinical phase | Number of patients | Intervention | Previous therapy | Results |
|---|---|---|---|---|---|
| Demetri et al., 2006 | III | 312 | Sunitinib 50 mg/d vs. placebo | Imatinib | mTTP: 6.3m vs. 1.5m, HR 0.33, p<.0001 mPFS: 5.5m vs. 1.4m, HR 0.33, p<.001 mOS: NR. HR 0.49, p=0.007 ORR: 7% vs. 0%, p<0.006 |
| Demetri et al., 2013 (GRID) | III | 199 | Regorafenib 160 mg/d vs. placebo | Imatinib and Sunitinib | mPFS: 4.8m vs. 0.9m, HR 0.27, p < 0.0001 HR OS: 0.77, p = 0.199 ORR: 4.5% vs. 1.5%, p<0.001 |
| Blay et al., 2020 (INVICTUS) | III | 129 | Ripretinib 150 mg/d vs. placebo | Imatinib, Suni- tinib and Regorafenib | mPFS: 6.3m vs. 1.0m, HR 0.15, p<0.0001 mOS: 15.1m vs. 6.6m, HR 0.36 ORR: 9% vs. 0%, p<0.054 |
| Mir et al., 2016 (PAZOGIST) | II | 81 | Pazopanib 800 mg/d + BSC vs. placebo + BSC | Imatinib and Sunitinib | mPFS: 3.4m vs. 2.3m, HR 0.59, p=0.03 mOS: 17.8m vs. 12.9m, HR 0.94, p=0.69 SD: 84% vs. 71% |
| Schöffski et al., 2020 (CaboGIST) | II | 50 | Cabozantinib 60 mg/d | Imatinib and Sunitinib | mPFS: 5.5m (95%IC, 3.6 - 6.9) mOS 18.2m (95%IC, 14.3 - 22.3) ORR: 14% |
| Park et al., 2012 | II | 31 | Sorafenib 800 mg/d | Imatinib and Sunitinib | mPFS: 4.9m (IC 95%, 1.3 - 8.5) mOS: 9.7m (IC 95%, 7.2 - 12.2) SD: 52%; PR: 13% |
| Reichardt et al., 2012 | III | 248 | Nilotinib 800 mg/d vs. BSC ± Imatinib or Sunitinib | Imatinib and Sunitinib | mPFS: 109d vs. 111d, HR 0.9, p=0.56 mOS: 332d vs. 280d, HR 0.79, p=0.29 CBR: 52.7% vs. 44.6%, p=0.28 |
Abbreviations: BCS = Best support of care; CBR = Clinical benefit rate; HR = Hazard ratio; ORR = Objective response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response; SD = Stable disease; TTP = Time to progression; d = Day; m = Months; y = Year.
In the phase III SWOG Intergroup trial S0033, 746 patients with advanced or unresectable GIST were randomized to receive 400 or 800 mg of imatinib daily. A higher dose of imatinib did not improve ORR, PFS or OS and was associated with increased toxicity. This study confirmed the standard daily dose of 400 mg imatinib as the preferred first-line treatment for advanced or unresectable GIST. In this study, 133 patients who experienced disease progression while on 400 mg daily imatinib were subsequently escalated to 800 mg daily; among the patients who were able to evaluate response after crossovers, 31% achieved either a partial response or stable disease.
On average, patients with exon 11 mutations experience disease progression within approximately 23 months, whereas those with exon 9 mutations have a median interval to progression of approximately 13 months.
Resistance to first-line therapy in GISTs can occur through various mechanisms, including secondary mutations in c-KIT (exons 13, 14, 17, and 18), RAS, BRAF, activation of alternative signaling pathways, c-KIT amplification, KIT-independent mechanisms of KIT phosphorylation, and efflux pump activity. (65) Most GISTs progress after an initial response to imatinib with the acquisition of chromosomal alterations, referred to as secondary resistance. The most common causes of secondary resistance in both KIT-mutant and PDGFRA-mutant GISTs are acquired cis-mutations in either the ATP-binding domain (encoded by exon 13 or 14 of KIT and exon 14 of PDGFRA) or the activation loop (encoded by exon 17 of KIT and exon 18 of PDGFRA). This distinction is crucial because GIST cells with acquired mutations in the ATP-binding domain have different sensitivities to other therapeutic options (e.g., sunitinib, regorafenib, and other TKIs).
In most studies, the frequency of primary mutations in exons 13 and 17 is 1-2%. Evidence regarding the efficacy of imatinib in these cases is still controversial and limited, with anecdotal reports of partial responses contradicted by more robust data describing resistance to imatinib in tumors harboring a secondary mutation in exon 17
Acquired secondary mutations are more prevalent in tumors with a primary exon 11 mutation and are less common in wild-type genotype GISTs, possibly due to the greater dependence of wildtype genotypes on KIT signaling. These secondary mutations confer resistance to imatinib and are commonly found in the ATP-binding domain (exons 13 and 14) or the KIT activation loop domain (exons 17 and 18).
Understanding the secondary mutations that confer imatinib resistance is important for prognostic implications and to guide subsequent treatment response. A meta-analysis of 1,083 GIST cases identified a prevalence of secondary mutations in 14% of cases, with 59% occurring in KIT and 3% in PDGFRA. The primary genotypes most prone to secondary mutations after first line imatinib treatment were those with exon 11 mutations (70%) and exon 9 mutations (39%). The most commonly reported secondary mutations in the KIT gene after treatment with imatinib are exon 17 mutations (55%), followed by exon 13 mutations (38%) and exon 14 mutations (13%). These findings highlight the importance of monitoring for secondary mutations and developing targeted therapies to overcome resistance in GIST patients who experience disease progression during imatinib treatment.
BRAF mutations in GISTs occur in approximately 4% of adult KIT/PDGFRA wild-type GISTs and are nearly mutually exclusive.
Sunitinib is a small molecule with multiple targets, including KIT, PDGFRA, RET, tyrosine kinase three associated with FMS, and colony stimulating factor 1 receptor (CSF1R), presenting antiangiogenic functionality by inhibiting vascular endothelial growth factor (VEGFR1-3). Validation of the drug as a second-line treatment for advanced GIST after failure or intolerance to imatinib was demonstrated in a phase III study in which 312 patients were randomized to receive 50 mg of sunitinib daily or placebo. The primary outcome of time to tumor progression (TTP) was more than four times superior to placebo at 27.3 weeks versus 6.4 weeks. (HR = 0.33, 95%CI = 0ꞏ23 to 047; p <0.0001), demonstrating a reduction in the risk of progression in all subgroups (all with HR<0.5). However, objective responses to second line sunitinib occurred in only 7% of patients (versus 0% in the placebo arm). The incidence of treatment-related toxicities of any grade was 83% and included myelotoxicity, fatigue, diarrhea, skin lesions and nausea. As a result of this randomized trial, sunitinib was approved by the FDA in 2006 for imatinib-refractory patients.
Although sunitinib is considered the standard second-line treatment according to current guidelines, limited clinical trials have shown the effectiveness of sunitinib in tumors with secondary KIT mutations occurring in the drug/ATP binding pocket (exon 13 and 14). At the same time, it is ineffective in tumors with localized resistance mutations in the activation loop (exon 17 and 18).
Regorafenib, a multikinase inhibitor that primarily targets KIT, PDGFR and VEGFR, was evaluated in the phase III GRID trial, the first clinical trial to demonstrate clinical benefit with a TKI in the post-imatinib and post-sunitinib setting. Regorafenib was compared to placebo, and resulted in a significant improvement in PFS (median PFS 4.8 months versus 0.9 months, respectively [HR = 0.27, 95%CI = 0.19 to 0.39; p<.0001]), regardless of KIT exon 9 and 11 mutations. However, there was no evidence of an improvement in OS, possibly due to a high crossover rate in the placebo arm.
The INVICTUS trial compared ripretinib, a switchcontrol TKI, to placebo for patients with GIST who progressed on imatinib, sunitinib, and regorafenib. Ripretinib demonstrated a significant improvement in PFS compared to placebo with 6.3 months versus 1 month, respectively (HR = 0.15, 95%CI = 0.09 to 0.25; p<0.0001) and had a favorable safety profile, leading to regulatory approvals as a fourth line therapy. Grade 3 or 4 adverse events with ripretinib included increased lipase levels (5%), as well as arterial hypertension (4%), and fatigue (2%). Further analysis also showed an improvement in quality of life.
Based on these encouraging results, the INTRIGUE trial evaluated ripretinib as a second-line therapy and directly compared it with sunitinib. Although ripretinib showed a numerical improvement in the primary outcome of PFS, the difference was not statistically significant at 8.3 months versus 7 months, respectively (HR = 0.88; 95%CI = 0.66 to 1.16; p=0.36). However, ripretinib resulted in greater clinical benefit in patients with KIT exon 11 and secondary exon 17/18 (activation loop) mutations, whereas those with secondary exon 13/14 (ATP-binding pocket) mutations had greater benefit from sunitinib.
There is currently no standardized approach to TKI selection in subsequent lines of GIST therapy. Retrospective studies, case series, or phase II clinical trials are the basis for most indications, showing limited clinical benefit in managing toxicity in patients with compromised clinical performance.
One TKI being evaluated in the advanced scenario is pazopanib, a multitarget inhibitor of KIT, VEGFR1-3, and PDGFR A-B. The phase II PAZOGIST trial compared pazopanib with best supportive care in 81 patients previously treated with imatinib and sunitinib. The intervention group demonstrated a longer PFS than the control group (3.4 months versus 2.3 months, p=0.03). However, the use of pazopanib was associated with considerable grade 3/4 adverse effects (72%), including systemic arterial hypertension (38%) and pulmonary embolism (13%).
Other treatment options for GISTs in later lines include cabozantinib (CABOGIST trial).
Sorafenib, a multikinase inhibitor targeting KIT, VEGFR, PDGFR-ß, and BRAF kinase, has gained a place in the treatment of advanced GIST, primarily in tumors resistantto imatinib and sunitinib that harbor localized resistance mutations in the activation loop (exons 17 and 18). A phase II study by the University of Chicago Consortium evaluated sorafenib in 38 imatinib- and sunitinib-resistant patients. It achieved a disease control rate (DCR) of 68% and a PFS of 5.2 months at the expense of some grade 3/4 adverse events such as hand-foot syndrome (45%), hypertension (21%) and diarrhea (8%). Data were similar to other cohorts, such as a Korean phase II study that found a DCR of 36% at 24 weeks and PFS of 4.9 months in 31 patients who failed two or more prior TKI, being significantly shorter in patients with primary genotypes other than KIT exon 11 mutation and with previous use of nilotinib.
To identify potential biomarkers predictive of response to sorafenib, three patients with a primary mutation in exon 11 of KIT who had developed resistance to imatinib, sunitinib and regorafenib were treated in a case series. The author showed that the patient with a secondary mutation in BRAF V600E had a better PFS with regorafenib and a worse PFS with sorafenib.
Nilotinib, another selective TKI, acts by inhibiting the tyrosine kinase activity of ABL1/BCR-ABL1 and KIT as well as PDGFRs and the discoidin domain receptor. Some trials evaluating this drug as a third-line or subsequent treatment option have yielded discouraging results, with a DCR of 37% and no superiority in PFS over best supportive care.
Regarding the SDH-mutant GIST population, a post hoc analysis of the S0033 trial, incorporating tumor DNA sequencing, revealed that 12 out of 20 cases with KIT/PDGFRA wt had mutated pathways in SDH. Additionally, this analysis identified a response rate of 8% with imatinib use in this population. This rate is significantly lower than the response rate observed in patients with KIT exon 11-mutant tumors, who exhibited a response rate of approximately 66% (p < .001).
Analysis of tumor DNA identified by ctDNA is of great interest in the management of GIST in various scenarios, such as early detection of relapse, evaluation of treatment response, and identification of new secondary mutations that may confer resistance to ongoing treatment.
Prior to initiating regorafenib in the GRID trial, investigators performed ctDNA analysis using the SafeSEQ technique. It showed that although there could be a potential association between high ctDNA levels and increased tumor burden, researchers did not observe a statistically significant association between baseline ctDNA levels and survival data. The timing of the biopsy, performed prior to regorafenib treatment, may have influenced this analysis. These findings highlight the impact that new sequencing methods may have on the management of GIST.
Researchers performed a liquid biopsy, analyzing 29 genes in a sample of46 patients during treatment for GIST at various stages. Of the ten patients who showed disease progression, seven had mutations detected in ctDNA at the time of measurable metastatic disease progression. These mutations were no longer detectable after clinical response to a new line of therapy. However, the authors emphasize that not all GISTs release ctDNA for mutation detection and that tumor burden during analysis may interfere with the detection of these mutations.
Another critical issue is the evaluation of germline alterations in GIST patients without known hereditary syndromes. By expanding genetic testing to 106 GIST samples, approximately 23% had pathogenic/likely pathogenic germline variants in genes associated with GIST and 8% had variants in other cancer susceptibility genes. The researchers identified GIST- associated gene variants (SDHA, SDHB, SDHC, NF1, KIT) in 69% of patients with KIT/PDGFRA wild-type GISTs, 63% of whom had no personal or family history of syndromic features. These findings underscore the importance of comprehensive genetic screening in GIST patients, even in the absence of syndromic features, which may have significant implications for diagnosis, prognosis, and potential personalized therapeutic interventions. Therefore, consideration of expanded genetic testing may be beneficial in optimizing patient management and tailored treatment approaches for GIST.
Advances in DNA and RNA sequencing methods have revealed novel, potentially actionable drivers in GIST. These discoveries include mutations in PIK3CA, overexpression of FGF4, and gene fusion proteins involving NTRK3 (ETV6-NTRK3) or FGFR1 (FGFR1- TACC1).
As the complexity of GIST treatment evolves because of an increasing number of agents, fortunately leading to improved outcomes, the incorporation of tools to better characterize the molecular basis of this disease offers a promising opportunity for rational treatment decisions. Such molecular insights promise to further revolutionize GIST treatment strategies, ultimately improving patient outcomes and paving the way for more personalized and effective therapeutic interventions.
NFLJ Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
MMQ Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
JSLF Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
DSRLJ Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
EFC Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
BMA Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
LGCAL Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
FT Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
FOF Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
EHA Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
FCC Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
RRM Collection and assembly of data, Conception and design, Data analysis and interpretation, Final approval of manuscript, Manuscript writing, Provision of study materials or patient.
| Genotype | Imatinib dose | No. of patients | Median TTP (months) | Median OS (months) |
|---|---|---|---|---|
| KIT exon 9 | 400 mg | 14 | 9.4 | 38.6 |
| 800 mg | 18 | 18 | 38.4 | |
| KIT exon 11 | 400 mg | 141 | 27.2 | 60.0 |
| 800 mg | 142 | 23.9 | NR | |
| Wild type | 400 mg | 43 | 15.6 | 49.0 |
| 800 mg | 24 | 9.8 | 39.5 |
Adapted from: Heinrich et al. (2008).
Abbreviations: TTP = Time to progression; OS = Overall survival; NR = Not reached; WT = Wild type.
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.
Journal: Brazilian Journal of Oncology
DOI: 10.1055/s-00059887
e-issn: 2526-8732
Publisher: Thieme Revinter Publicações Ltda.
Publisher address: Rua do Matoso 170, Rio de Janeiro, RJ, CEP 20270-135, Brazil
No citations found for this article.
1. Søreide, K and Sandvik, OM and Søreide, JA and Giljaca, V and Jureckova, A and Bulusu, VR. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol [online]. 2016, vol. 40, p. 39-46.
2. Parab, TM and DeRogatis, MJ and Boaz, AM and Grasso, SA and Issack, PS and Duarte, DA. Gastrointestinal stromal tumors: a comprehensive review. J Gastrointest Oncol [online]. 2018, vol. 10, p. 144-54.
3. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol [online]. 1983, vol. 7, p. 507-20.
4. Patel, N and Benipal, B. Incidence of Gastrointestinal Stromal Tumors in the United States from 2001-2015: A United States Cancer Statistics Analysis of 50 States. Cureus [online]. 2019, vol. 11, p. e4120.
5. Stanek, M and Pisarska, M and Budzyńska, D and Rzepa, A and Pędziwiatr, M and Major, P. Gastric gastrointestinal stromal tumors: clinical features and short- and longterm outcomes of laparoscopic resection. Wideochir Inne Tech Maloinwazyjne [online]. 2019, vol. 14, p. 176-81.
6. Cajal, SR. Sobre la existencia de células nerviosas especiales en la primera capa de las circunvoluciones cerebrales. Gaceta Médica Catalana [online]. 1890, vol. 13, p. 737-9.
7. Hirota, S and Isozaki, K and Moriyama, Y and Hashimoto, K and Nishida, T and Ishiguro, S. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science [online]. 1998, vol. 279, p. 577-80.
8. Graadt van Roggen, JF and van Velthuysen, ML and Hogendoorn, PC. The histopathological differential diagnosis of gastrointestinal stromal tumours. J Clin Pathol [online]. 2001, vol. 54, p. 96-102.
9. Shi, E and Chmielecki, J and Tang, CM and Wang, K and Heinrich, MC and Kang, G. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med [online]. 2016, vol. 14, p. 339.
10. Güler, B and Özyılmaz, F and Tokuç, B and Can, N and Taştekin, E. Histopathological Features of Gastrointestinal Stromal Tumors and the Contribution of DOG1 Expression to the Diagnosis. Balkan Med J [online]. 2015, vol. 32, p. 388-96.
11. Lech, G and Korcz, W and Kowalczyk, E and Guzel, T and Radoch, M and Krasnodębski, IW. Giant gastrointestinal stromal tumour of rare sarcomatoid epithelioid subtype: case study and literature review. World J Gastroenterol [online]. 2015, vol. 21, p. 3388-93.
12. Corless, CL. Gastrointestinal stromal tumors: what do we know now?. Mod Pathol [online]. 2014, vol. 27, p. S1-16.
13. Suresh, PK and Sahu, KK and Pai, RR and Sridevi, HB and Ballal, K and Khandelia, B. The Prognostic Significance of Neuroendocrine Differentiation in Colorectal Carcinomas: Our Experience. J Clin Diagn Res [online]. 2015, vol. 9, p. EC01-4.
14. Huizinga, JD and Thuneberg, L and Klüppel, M and Malysz, J and Mikkelsen, HB and Bernstein, A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature [online]. 1995, vol. 373, p. 347-9.
15. Cioffi, A and Maki, RG. GI Stromal Tumors: 15 Years of Lessons From a Rare Cancer. J Clin Oncol [online]. 2015, vol. 33, p. 1849-54.
16. Venkataraman, V and George, S and Cote, GM. Molecular Advances in the Treatment of Advanced Gastrointestinal Stromal Tumor. Oncologist [online]. 2023, vol. 28, p. 671-681.
17. Martín, J and Poveda, A and Llombart-Bosch, A and Ramos, R and López-Guerrero, JA and García del Muro, J. Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol [online]. 2005, vol. 23, p. 6190-8.
18. Wardelmann, E and Hrychyk, A and Merkelbach-Bruse, S and Pauls, K and Goldstein, J and Hohenberger, P. Association of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J Mol Diagn [online]. 2004, vol. 6, p. 197-204.
19. Bannon, AE and Klug, LR and Corless, CL and Heinrich, MC. Using molecular diagnostic testing to personalize the treatment of patients with gastrointestinal stromal tumors. Expert Rev Mol Diagn [online]. 2017, vol. 17, p. 445-57.
20. Moosavi, B and Zhu, XL and Yang, WC and Yang, GF. Molecular pathogenesis of tumorigenesis caused by succinate dehydrogenase defect. Eur J Cell Biol [online]. 2020, vol. 99, p. 151057.
21. Nannini, M and Biasco, G and Astolfi, A and Urbini, M and Pantaleo, MA. Insulin-like Growth Factor (IGF) system and gastrointestinal stromal tumours (GIST): present and future. Histol Histopathol [online]. 2014, vol. 29, p. 167-75.
22. Italiano, A and Chen, CL and Sung, YS and Singer, S and DeMatteo, RP and LaQuaglia, MP. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer [online]. 2012, vol. 12, p. 408.
23. Boikos, SA and Pappo, AS and Killian, JK and LaQuaglia, MP and Weldon, CB and George, S. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol [online]. 2016, vol. 2, p. 922-8.
24. Blay, JY and Kang, YK and Nishida, T and von Mehren, M. Gastrointestinal stromal tumours. Nat Rev Dis Primers [online]. 2021, vol. 7, p. 22.
25. Agaram, NP and Wong, GC and Guo, T and Maki, RG and Singer, S and DeMatteo, RP. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer [online]. 2008, vol. 47, p. 853-9.
26. Napolitano, A and Ostler, AE and Jones, RL and Huang, PH. Fibroblast Growth Factor Receptor (FGFR) Signaling in GIST and Soft Tissue Sarcomas. Cells [online]. 2021, vol. 10, p. 1533.
27. Nishida, T and Tsujimoto, M and Takahashi, T and Hirota, S and Blay, JY and Wataya-Kaneda, M. Gastrointestinal stromal tumors in Japanese patients with neurofibromatosis type I. J Gastroenterol [online]. 2016, vol. 51, p. 571-8.
28. DeMatteo, RP and Lewis, JJ and Leung, D and Mudan, SS and Woodruff, JM and Brennan, MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg [online]. 2000, vol. 231, p. 51-8.
29. Fletcher, CD and Berman, JJ and Corless, C and Gorstein, F and Lasota, J and Longley, BJ. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol [online]. 2002, vol. 33, p. 459-65.
30. Joensuu, H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol [online]. 2008, vol. 39, p. 1411-9.
31. Gold, JS and Gönen, M and Gutiérrez, A and Broto, JM and García-del-Muro, X and Smyrk, TC. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localised primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol [online]. 2009, vol. 10, p. 1045-52.
32. Rossi, S and Gasparotto, D and Miceli, R and Toffolatti, L and Gallina, G and Scaramel, E. KIT, PDGFRA, and BRAF mutational spectrum impacts on the natural history of imatinibnaive localized GIST: a population-based study. Am J Surg Pathol [online]. 2015, vol. 39, p. 922-30.
33. Clinicopathological Features and Surgical Management of Gastrointestinal Stromal Tumors: State-of-the-Art. Exon Publications, 2022.
34. Nishida, T and Blay, JY and Hirota, S and Kitagawa, Y and Kang, YK. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer [online]. 2016, vol. 19, p. 3-14.
35. Winder, A and Strauss, DC and Jones, RL and Benson, C and Messiou, C and Chaudry, MA. Robotic surgery for gastric gastrointestinal stromal tumors: A single center case series. J Surg Oncol [online]. 2020, vol. 122, p. 691-8.
36. Casali, PG and Abecassis, N and Aro, HT and Bauer, S and Biagini, R and Bielack, S. Corrections to “Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up”. Ann Oncol [online]. 2018, vol. 29, p. iv68-iv78.
37. von Mehren, M and Randall, RL and Benjamin, RS and Boles, S and Bui, MM and Ganjoo, KN. Soft Tissue Sarcoma, Version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw [online]. 2018, vol. 16, p. 536-63.
38. Mayr, P and Märkl, B and Agaimy, A and Kriening, B and Dintner, S and Schenkirsch, G. Malignancies associated with GIST: a retrospective study with molecular analysis of KIT and PDGFRA. Langenbecks Arch Surg [online]. 2019, vol. 404, p. 605-13.
39. Iwatsuki, M and Harada, K and Iwagami, S and Eto, K and Ishimoto, T and Baba, Y. Neoadjuvant and adjuvant therapy for gastrointestinal stromal tumors. Ann Gastroenterol Surg [online]. 2018, vol. 3, p. 43-49.
40. Dematteo, RP and Ballman, KV and Antonescu, CR and Maki, RG and Pisters, PW and Demetri, GD. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet [online]. 2009, vol. 373, p. 1097-104.
41. Corless, CL and Ballman, KV and Antonescu, CR and Kolesnikova, V and Maki, RG and Pisters, PW. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol [online]. 2014, vol. 32, p. 1563-70.
42. Casali, PG and Le Cesne, A and Poveda Velasco, A and Kotasek, D and Rutkowski, P and Hohenberger, P. Time to Definitive Failure to the First Tyrosine Kinase Inhibitor in Localized GI Stromal Tumors Treated With Imatinib As an Adjuvant: A European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Intergroup Randomized Trial in Collaboration With the Australasian Gastro-Intestinal Trials Group, UNICANCER, French Sarcoma Group, Italian Sarcoma Group, and Spanish Group for Research on Sarcomas. J Clin Oncol [online]. 2015, vol. 33, p. 4276-83.
43. Casali, PG and Le Cesne, A and Poveda, A and Kotasek, D and Rutkowski, P and Hohenberger, P. Time to definitive failure to the first tyrosine kinase inhibitor in localized gastrointestinal stromal tumors (GIST) treated with imatinib as an adjuvant: Final results of the EORTC STBSG, AGITG, UNICANCER, FSG, ISG, and GEIS randomized trial. Ann Oncol [online]. 2017, vol. 28, p. v645.
44. Rutkowski, P and Przybył, J and Zdzienicki, M. Extended adjuvant therapy with imatinib in patients with gastrointestinal stromal tumors: recommendations for patient selection, risk assessment, and molecular response monitoring. Mol Diagn Ther [online]. 2013, vol. 17, p. 9-19.
45. Joensuu, H and Eriksson, M and Sundby Hall, K and Hartmann, JT and Pink, D and Schütte, J. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA [online]. 2012, vol. 307, p. 1265-72.
46. Joensuu, H and Eriksson, M and Sundby Hall, K and Reichardt, A and Hermes, B and Schütte, J. Survival Outcomes Associated With 3 Years vs 1 Year of Adjuvant Imatinib for Patients With High-Risk Gastrointestinal Stromal Tumors: An Analysis of a Randomized Clinical Trial After 10-Year Follow-up. JAMA Oncol [online]. 2020, vol. 6, p. 1241-6.
47. Chibon, F and Lagarde, P and Salas, S and Pérot, G and Brouste, V and Tirode, F. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat Med [online]. 2010, vol. 16, p. 781-7.
48. Raut, CP and Espat, NJ and Maki, RG and Araujo, DM and Trent, J and Williams, TF. Efficacy and Tolerability of 5-Year Adjuvant Imatinib Treatment for Patients With Resected Intermediate- or High-Risk Primary Gastrointestinal Stromal Tumor: The PERSIST-5 Clinical Trial. JAMA Oncol [online]. 2018, vol. 4, p. e184060.
49. ClinicalTrials.gov [Internet]. National Library of Medicine (US), 2000.
50. ClinicalTrials.gov [Internet]. National Library of Medicine (US), 2014.
51. Heinrich, MC and Corless, CL and Demetri, GD and Blanke, CD and von Mehren, M and Joensuu, H. Kinase Mutations and Imatinib Response in Patients With Metastatic Gastrointestinal Stromal Tumor. J Clin Oncol [online]. 2023, vol. 41, p. 4829-36.
52. Ishikawa, T and Kanda, T and Kameyama, H and Wakai, T. Neoadjuvant therapy for gastrointestinal stromal tumor. Transl Gastroenterol Hepatol [online]. 2018, vol. 3, p. 3.
53. von Mehren, M and Joensuu, H. Gastrointestinal Stromal Tumors. J Clin Oncol [online]. 2018, vol. 36, p. 136-43.
54. Blanke, CD and Demetri, GD and von Mehren, M and Heinrich, MC and Eisenberg, B and Fletcher, JA. Long-term results from a randomized phase II trial of standardversus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol [online]. 2008, vol. 26, p. 620-5.
55. Tirumani, SH and Shinagare, AB and Jagannathan, JP and Krajewski, KM and Ramaiya, NH and Raut, CP. Radiologic assessment of earliest, best, and plateau response of gastrointestinal stromal tumors to neoadjuvant imatinib prior to successful surgical resection. Eur J Surg Oncol [online]. 2014, vol. 40, p. 420-8.
56. Mar 13;(Version 1.2023). Available from [online]. Available from: <https://www.nccn.org/professionals/physician_gls/pdf/gist.pdf>.
57. Van den Abbeele, AD and Gatsonis, C and de Vries, DJ and Melenevsky, Y and Szot-Barnes, A and Yap, JT. ACRIN 6665/RTOG 0132 phase II trial of neoadjuvant imatinib mesylate for operable malignant gastrointestinal stromal tumor: monitoring with 18F-FDG PET and correlation with genotype and GLUT4 expression. J Nucl Med [online]. 2012, vol. 53, p. 567-74.
58. Farag, S and IJzerman, NS and Houdijk, MPM and Reyners, AKL and Arens, AI and Grünhagen, DJ. Early response evaluation using 18F-FDG-PET/CT does not influence management of patients with metastatic gastrointestinal stromal tumors (GIST) treated with palliative intent. Nuklearmedizin [online]. 2021, vol. 60, p. 411-6.
59. Demetri, GD and von Mehren, M and Blanke, CD and Van den Abbeele, AD and Eisenberg, B and Roberts, PJ. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med [online]. 2002, vol. 347, p. 472-80.
60. von Mehren, M and Heinrich, MC and Joensuu, H and Blanke, CD and Wehrle, E and Demetri, GD. Follow-up results after 9 years (yrs) of the ongoing, phase II B2222 trial of imatinib mesylate (IM) in patients (pts) with metastatic or unresectable KIT+ gastrointestinal stromal tumors (GIST). J Clin Oncol [online]. 2011, vol. 29, p. 10016.
61. Blanke, CD and Rankin, C and Demetri, GD and Ryan, CW and von Mehren, M and Benjamin, RS. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol [online]. 2008, vol. 26, p. 626-32.
62. Verweij, J and Casali, PG and Zalcberg, J and LeCesne, A and Reichardt, P and Blay, JY. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet [online]. 2004, vol. 364, p. 1127-34.
63. Zalcberg, JR and Verweij, J and Casali, PG and Le Cesne, A and Reichardt, P and Blay, JY. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer [online]. 2005, vol. 41, p. 1751-7.
64. Casali, PG and Zalcberg, J and Le Cesne, A and Reichardt, P and Blay, JY and Lindner, LH. Ten-Year Progression-Free and Overall Survival in Patients With Unresectable or Metastatic GI Stromal Tumors: Long-Term Analysis of the European Organisation for Research and Treatment of Cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group Intergroup Phase III Randomized Trial on Imatinib at Two Dose Levels. J Clin Oncol [online]. 2017, vol. 35, p. 1713-20.
65. Debiec-Rychter, M and Sciot, R and Le Cesne, A and Schlemmer, M and Hohenberger, P and van Oosterom, AT. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer [online]. 2006, vol. 42, p. 1093-103.
66. Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of Two Doses of Imatinib for the Treatment of Unresectable or Metastatic Gastrointestinal Stromal Tumors: A Meta-Analysis of 1,640 Patients. J Clin Oncol [online]. 2010, vol. 28, p. 1247-53.
67. Heinrich, MC and Owzar, K and Corless, CL and Hollis, D and Borden, EC and Fletcher, CD. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol [online]. 2008, vol. 26, p. 5360-7.
68. Heinrich, MC and Corless, CL and Blanke, CD and Demetri, GD and Joensuu, H and Roberts, PJ. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol [online]. 2006, vol. 24, p. 4764-74.
69. Nishida, T and Kanda, T and Nishitani, A and Takahashi, T and Nakajima, K and Ishikawa, T. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib‐resistant gastrointestinal stromal tumor. Cancer Sci [online]. 2008, vol. 99, p. 799-804.
70. Demetri, GD and van Oosterom, AT and Garrett, CR and Blackstein, ME and Shah, MH and Verweij, J. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet [online]. 2006, vol. 368, p. 1329-38.
71. Demetri, GD and Reichardt, P and Kang, YK and Blay, JY and Rutkowski, P and Gelderblom, H. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet [online]. 2013, vol. 381, p. 295-302.
72. Wozniak, A and Rutkowski, P and Piskorz, A and Ciwoniuk, M and Osuch, C and Bylina, E. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann Oncol [online]. 2012, vol. 23, p. 353-60.
73. Kang, G and Lee, J and Jang, KT and Beadling, C and Corless, CL and Heinrich, MC. Multiplex mutation screening by mass spectrometry in gastrointestinal stromal tumours. Pathology [online]. 2012, vol. 44, p. 460-4.
74. Ko, TK and Lee, E and Ng, CCY and Yang, VS and Farid, M and Teh, BT. Circulating Tumor DNA Mutations in Progressive Gastrointestinal Stromal Tumors Identify Biomarkers of Treatment Resistance and Uncover Potential Therapeutic Strategies. Front Oncol [online]. 2022, vol. 12, p. 840843.
75. Heinrich, MC and Maki, RG and Corless, CL and Antonescu, CR and Harlow, A and Griffith, D. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol [online]. 2008, vol. 26, p. 5352-9.
76. Lee, JH and Kim, Y and Choi, JW and Kim, YS. Correlation of imatinib resistance with the mutational status of KIT and PDGFRA genes in gastrointestinal stromal tumors: a meta-analysis. J Gastrointestin Liver Dis [online]. 2013, vol. 22, p. 413-8.
77. Huss, S and Pasternack, H and Ihle, MA and Merkelbach-Bruse, S and Heitkötter, B and Hartmann, W. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol [online]. 2017, vol. 62, p. 206-214.
78. My Cancer Genome®. Gastrointestinal Stromal Tumor [Data set]. Vanderbilt University, Vanderbilt-Ingram Cancer Center, 2015.
79. Astolfi, A and Pantaleo, MA and Indio, V and Urbini, M and Nannini, M. The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. Int J Mol Sci [online]. 2020, vol. 21, p. 3313.
80. Gasparotto, D and Rossi, S and Polano, M and Tamborini, E and Lorenzetto, E and Sbaraglia, M. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin Cancer Res [online]. 2017, vol. 23, p. 273-82.
81. Joensuu, H and Blay, JY and Comandone, A and Martin-Broto, J and Fumagalli, E and Grignani, G. Dovitinib in patients with gastrointestinal stromal tumour refractory and/or intolerant to imatinib. Br J Cancer [online]. 2017, vol. 117, p. 1278-85.
82. Soria, JC and Massard, C and Magné, N and Bader, T and Mansfield, CD and Blay, JY. Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib in advanced and/or metastatic solid cancers. Eur J Cancer [online]. 2009, vol. 45, p. 2333-41.
83. Prenen, H and Cools, J and Mentens, N and Folens, C and Sciot, R and Schöffski, P. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res [online]. 2006, vol. 12, p. 2622-7.
84. Jeffers, M and Kappeler, C and Kuss, I and Beckmann, G and Mehnert, DH and Fredebohm, J. Broad spectrum of regorafenib activity on mutant KIT and absence of clonal selection in gastrointestinal stromal tumor (GIST): correlative analysis from the GRID trial. Gastric Cancer [online]. 2022, vol. 25, p. 598-608.
85. Blay, JY and Serrano, C and Heinrich, MC and Zalcberg, J and Bauer, S and Gelderblom, H. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebocontrolled, phase 3 trial. Lancet Oncol [online]. 2020, vol. 21, p. 923-34.
86. Bauer, S and Jones, RL and Blay, JY and Gelderblom, H and George, S and Schöffski, P. Ripretinib Versus Sunitinib in Patients With Advanced Gastrointestinal Stromal Tumor After Treatment With Imatinib (INTRIGUE): A Randomized, Open-Label, Phase III Trial. J Clin Oncol [online]. 2022, vol. 40, p. 3918-28.
87. Mir, O and Cropet, C and Toulmonde, M and Cesne, AL and Molimard, M and Bompas, E. Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): a randomised, multicentre, open-label phase 2 trial. Lancet Oncol [online]. 2016, vol. 17, p. 632-41.
88. Eriksson, M and Reichardt, P and Joensuu, H and Krarup-Hansen, A and Hagberg, O and Hohenberger, P. Benefit of pazopanib in advanced gastrointestinal stromal tumours: results from a phase II trial (SSG XXI, PAGIST). ESMO Open [online]. 2021, vol. 6, p. 100217.
89. Schöffski, P and Mir, O and Kasper, B and Papai, Z and Blay, JY and Italiano, A. Activity and safety of the multitarget tyrosine kinase inhibitor cabozantinib in patients with metastatic gastrointestinal stromal tumour after treatment with imatinib and sunitinib: European Organisation for Research and Treatment of Cancer phase II trial 1317 ‘CaboGIST’. Eur J Cancer [online]. 2020, vol. 134, p. 62-74.
90. Park, SH and Ryu, MH and Ryoo, BY and Im, SA and Kwon, HC and Lee, SS. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs [online]. 2012, vol. 30, p. 2377-83.
91. Montemurro, M and Gelderblom, H and Bitz, U and Schütte, J and Blay, JY and Joensuu, H. Sorafenib as third- or fourthline treatment of advanced gastrointestinal stromal tumour and pretreatment including both imatinib and sunitinib, and nilotinib: A retrospective analysis. Eur J Cancer [online]. 2013, vol. 49, p. 1027-31.
92. Heinrich, MC and Marino-Enriquez, A and Presnell, A and Donsky, RS and Griffith, DJ and McKinley, A. Sorafenib inhibits many kinase mutations associated with drugresistant gastrointestinal stromal tumors. Mol Cancer Ther [online]. 2012, vol. 11, p. 1770-80.
93. Franck, C and Rosania, R and Franke, S and Haybaeck, J and Canbay, A and Venerito, M. The BRAF Status May Predict Response to Sorafenib in Gastrointestinal Stromal Tumors Resistant to Imatinib, Sunitinib, and Regorafenib: Case Series and Review of the Literature. Digestion [online]. 2019, vol. 99, p. 179-84.
94. Reichardt, P and Blay, JY and Gelderblom, H and Schlemmer, M and Demetri, GD and Bui-Nguyen, B. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann Oncol [online]. 2012, vol. 23, p. 1680-7.
95. Selecting tyrosine kinase inhibitors for gastrointestinal stromal tumor with secondary KIT activation-loop domain mutations. PLoS One [online]. 2013, vol. 8, p. e65762.
96. Spitaleri, G and Biffi, R and Barberis, M and Fumagalli, C and Toffalorio, F and Catania, C. Inactivity of imatinib in gastrointestinal stromal tumors (GISTs) harboring a KIT activation-loop domain mutation (exon 17 mutation pN822K). Onco Targets Ther [online]. 2015, vol. 8, p. 1997-2003.
97. Blay, JY and Shen, L and Kang, YK and Rutkowski, P and Qin, S and Nosov, D. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): a randomised phase 3 trial. Lancet Oncol [online]. 2015, vol. 16, p. 550-60.
98. Heinrich, MC and Rankin, C and Blanke, CD and Demetri, GD and Borden, EC and Ryan, CW. Correlation of Long-term Results of Imatinib in Advanced Gastrointestinal Stromal Tumors With Next-Generation Sequencing Results: Analysis of Phase 3 SWOG Intergroup Trial S0033. JAMA Oncol [online]. 2017, vol. 3, p. 944-52.
99. Janeway, KA and Kim, SY and Lodish, M and Nosé, V and Rustin, P and Gaal, J. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A [online]. 2011, vol. 108, p. 314-8.
100. von Mehren, M and George, S and Heinrich, MC and Schuetze, SM and Yap, JT and Yu, JQ. Linsitinib (OSI-906) for the Treatment of Adult and Pediatric Wild-Type Gastrointestinal Stromal Tumors, a SARC Phase II Study. Clin Cancer Res [online]. 2020, vol. 26, p. 1837-45.
101. Jilg, S and Rassner, M and Maier, J and Waldeck, S and Kehl, V and Follo, M. Circulating cKIT and PDGFRA DNA indicates disease activity in Gastrointestinal Stromal Tumor (GIST). Int J Cancer [online]. 2019, vol. 145, p. 2292-303.
102. Mandelker, D and Marra, A and Mehta, N and Selenica, P and Yelskaya, Z and Yang, C. Expanded genetic testing of GIST patients identifies high proportion of non-syndromic patients with germline alterations. NPJ Precis Oncol [online]. 2023, vol. 7, p. 1.
103. Cocco, E and Scaltriti, M and Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol [online]. 2018, vol. 15, p. 731-47.
104. Serrano, C and Wang, Y and Mariño-Enríquez, A and Lee, JC and Ravegnini, G and Morgan, JA. KRAS and KIT Gatekeeper Mutations Confer Polyclonal Primary Imatinib Resistance in GI Stromal Tumors: Relevance of Concomitant Phosphatidylinositol 3-Kinase/AKT Dysregulation. J Clin Oncol [online]. 2015, vol. 33, p. e93-6.
105. Urbini, M and Astolfi, A and Indio, V and Nannini, M and Schipani, A and Bacalini, MG. Gene duplication, rather than epigenetic changes, drives FGF4 overexpression in KIT/PDGFRA/SDH/RAS-P WT GIST. Sci Rep [online]. 2020, vol. 10, p. 19829.
Dados de acesso insuficientes para visualização no mapa.