Ewing's sarcoma (ES) is a family of neoplasms arising from ectopic neural and neuroectodermal tissues, first described in 1921. The extraosseous Ewing's sarcoma/ primitive neuroectodermal tumor (EWS/PNET) form was documented in 1969.
One of the subtypes of this family is the peripheral primitive neuroectodermal tumor (pPNET). This extremely rare disease affects mainly children, adolescents and young adults.
The prognosis of peripheral primitive neuroectodermal tumor (pPNET) is poor and early detection, complete surgical resection, chemotherapy and radiotherapy are considered standard care.
A few more than 30 cases of neuroectodermal primitive tumor originating on pancreas have been documented in the consulted literature. We would like to add a report of a locally advanced pancreatic pPNET treated by European Guidelines at the time, with the longest follow- up, of 8 years, considering quality of life outcomes. The patient signed an informed consent form and agreed to the elaboration of this report.
A 22-year-old, male, previously healthy patient presented pain, precocious gastric fullness, abdominal mass and 10kg weight loss for 3 months; imaging exams showed a tumor located in the body and tail of the pancreas about 14cm and gastric invasion (
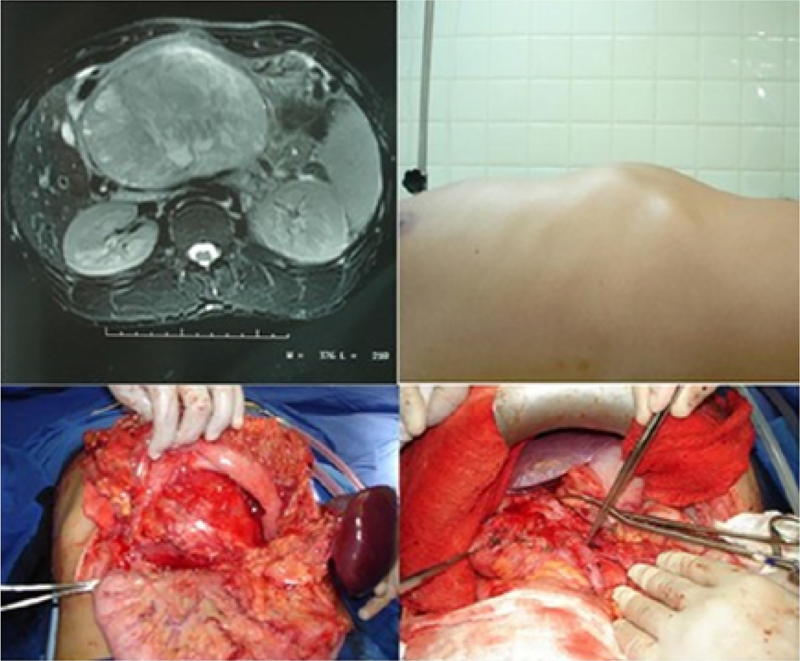
Figure 1 Upper left: abdomen CT showing a large tumor in pancreatic body and tail, with approximately 14cm in diameter; Upper right: profile photograph of the abdomen showing tumor in epigastrium; Left and right lower: photographs showing pancreatic tail tumor after retrocavity approach (left) and ligation of the splenic vein in the portal vein formation (right).
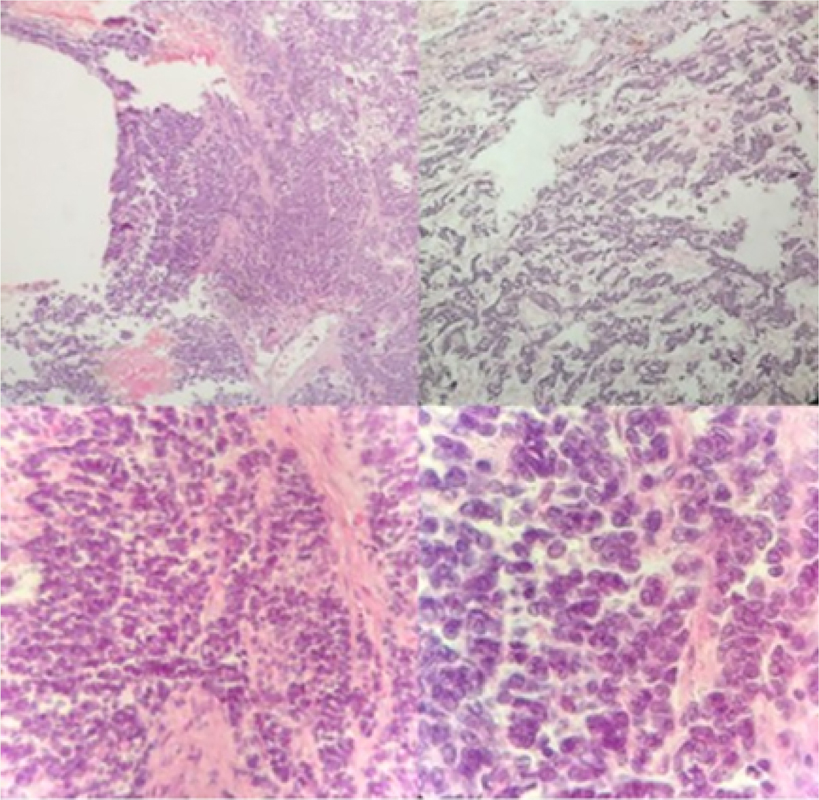
Post-operatory histopathologic findings were compatible with poorly differentiated malignant neoplasm with extensive small round cell components. Immunocytochemistry showed CD99 positivity, sarcomeric actin, pan-cytokeratin (AE1-AE3) and neuron-specific enolase strongly suggesting the diagnosis of EWS/PNET sarcoma (
Figure 2 Histopathology on hematoxylin-eosin stain. Upper left: 4x objective; Upper right: 10x objective; Lower left: 40x objective; Lower right: 100x objective.
| Domain | Results |
|---|---|
| Physical Functioning (PF) | 95 |
| Role-Physical (RP) | 75 |
| Bodily Pain (BP) | 100 |
| General Health (GH) | 42 |
| Vitality (VT) | 50 |
| Social Functioning (SF) | 100 |
| Role-Emotional (RE) | 100 |
| Mental Health (MH) | 72 |
Ewing's sarcoma/primitive neuroectodermal tumor (EWS/PNET) consists of four sub- types of tumors: Ewing's sarcoma of bone (ESB), extraosseous Ewing's sarcoma (EES), a peripheral primitive neuroectodermal tumor (pPNET) and Askin's tumor.
In the United States, there are an estimated 13,040 new cases of soft tissue sarcoma and 5,150 deaths due to this type of neoplasia in 2018, of which 1% are EWS/PNET.
The poor prognosis associated with these tumors is associated with its aggressive behavior with a tendency to recurrence and metastasis, already reported to lung, bone, bone marrow, liver and lymph nodes.
The diagnosis of EWS/PNET requires the association of diverse information. The suspicious of this tumor should be led by epidemiological data, especially considering a young age when most of the cases are diagnosed and clinical history and examination, which can be unspecific, compatible with growth of the tumor.
Imaging exams such as computerized tomography (CT), magnetic resonance imaging (MRI) and ultrasonography (US), also do not show radiologic characteristic signs. However, most tumors are encapsulated, hypoechoic on US, hypodense on CT, iso-hypointense on T1 and hyperintense on T2 on MRI.
The histomorphology of ES/PNET shows a poorly differentiated tumor of small round cells requiring a differential diagnosis with several other tumors including lymphoma, Wilms' tumor, neuroblastoma, pancreatoblastoma, pancreatic endocrine tumor, sarcoma and neuroendocrine carcinoma.
The therapeutic approach of patients with tumors of the Ewing's sarcoma family requires local control of the disease associated with systemic treatment.
Complete resection, including tumor-free margins, predicts gain on survival and control of local disease.
EES are radiation sensible, but risks of developing secondary malignancies and advances in surgical techniques restrict its indication. Radiotherapy is mostly indicated in cases of unresectable tumors and should be chosen as the only local control treatment on these cases, since its combination with intralesional resection did not show effectiveness. Neoadjuvant radiotherapy is recommended for locally advanced disease that suggests the surgical approach may leave residual disease.
The inclusion of multi-agent chemotherapy is associated with about 50-60% of improvement in long-term survival. The drugs that constitute the basic scheme in the protocols of most specialized centers are vincristine, cyclophosphamide, doxorubicin, ifosfamide, etoposide and dactinomycin,
Due to EWS/PNET aggressiveness and its subsequent hostile therapeutic, there may be a negative impact on the quality of life of patients treated. Quality of life was documented with the Medical Outcomes Study Questionnaire (SF - 36), which consists of 36 items grouped in 8 domains: physical functioning, role-physical, bodily pain, general health, vitality, social functioning, role-emotional and mental health. Scores values of the 8 domains are calculated, ranging from 0 being the worst to 100 as the best result, for each domain. In the case presented, only two domains (general health and vitality) tended to average scores resulting in great overall quality of life results. Thus, we suggest the adequate treatment provided beneficiated not only survival but also allowed a full life with social relations, without pain and little physical and emotional limitation, according to the patient's perception. Also, the report of QoL provided by the own patient highlights the patients' protagonism during the therapies implemented.
In conclusion, there have been described in the consulted literature a few more than 30 cases of primitive neuroectodermal tumors of pancreatic origin. In pancreatic neoplasms, primitive neuroectodermal tumors present great diagnostic challenge, and, although rare, should be part of the differential diagnosis of a pancreatic tumor in young patients. The aggressive surgical approach associated with multimodal regimens provided improvement in disease-free survival and a positive impact on the quality of life many years after treatment. The limitation of our conclusion consists of the own case report studies bias.
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.
Journal: Brazilian Journal of Oncology
DOI: 10.1055/s-00059887
e-issn: 2526-8732
Publisher: Thieme Revinter Publicações Ltda.
Publisher address: Rua do Matoso 170, Rio de Janeiro, RJ, CEP 20270-135, Brazil
No references with the required fields found.
Dados de acesso insuficientes para visualização no mapa.