The high cost of cancer drugs, especially in developing countries, makes it difficult for patients to access the best available treatment. With new generic options for this product, it is expected that prices will fall back to more accessible levels and thus provide an improvement in the quality of health of users. Studies have shown that the inclusion of generic drugs in the treatment of CML improves patient access to treatment. Generic medicines tend to cost less than their brand-name counterparts because they do not have to repeat animal and clinical (human) studies that were required of the brand-name medicines to demonstrate safety and effectiveness. Lower-cost generic drugs have been shown to increase the likelihood that patients take essential medications prescribed by their doctors and to improve patients’ health outcomes.
Two formulations, typically test and reference, are considered bioequivalent, if the ranges are between 80% and 125% according to Anvisa’s Resolution RE number 1170, dated April 19th, 2006,[1] which determines that two drugs will be considered bioequivalent if the extreme values of the 90% confidence interval of the ratio of geometric means (AUC0-t test/AUC0-t reference and Cmax test/Cmax reference) are greater than 80% and less than 125%.
Lenalidomide (IUPAC 3-(7-amino-3-oxo-1H-isoindol-2-yl)piperidine-2,6-dione), a thalidomide analogue, is an immunomodulatory (IMiD) and antineoplastic agent used in multiple myeloma therapy.
In 2005, the FDA approved the marketing of 5mg and 10mg Revlimid® (Lenalidomide) capsules for the treatment of transfusion-depending anemia patients due to low risk myelodysplastic syndrome (MDS) associated to 5q deletion abnormality with or without additional cytogenetic abnormalities and multiple myeloma in combination with dexamethasone.
In 2020, Anvisa issued RDC 393/2020, which included lenalidomide therapeutic indications for the treatment of multiple myeloma, in combination with bortezomib and dexamethasone, for patients with no prior treatment; for the treatment of follicular lymphoma or previously treated patients with marginal zone lymphoma, in combination with rituximab (anti-CD20 antibody), and relapsed/refractory mantle cell lymphoma.
As to the security profile, authors report that lenalidomide is well tolerated and that most usual adverse events include hematologic toxicity with controllable neutropenia and thrombocytopenia.
The objective of this study was to verify whether the rate and extent of absorption of the 10mg lenalidomide immediate release capsule formulation manufactured by Eurofarma Laboratórios S.A. are equivalent to those of the reference product, Revlimid®, when administered in one single-dose and under fasting conditions to adult healthy male subjects.
The test drug - 10mg lenalidomide immediate release capsule -, was manufactured by Eurofarma Laboratórios S/A. The reference product used in the study was Revlimid® (10mg lenalidomide immediate release capsule), manufactured by Celgene Europe B.V. and registered in Brazil by Bristol-Myers Squibb Farmacêutica LTDA.
Based on its mechanism of action and findings from animal studies, lenalidomide can cause embryofetal harm when administered to a pregnant female and is contraindicated during pregnancy.
Thirty-two Mexican adult male healthy volunteers willing to participate in the study were selected based on the protocol eligibility criteria 90 days before to the first study period. A sufficient number of eligible volunteers showed up in the research center facilities, and the 32 volunteers who fulfilled the protocol requirements were given information regarding the study and, after having their inquiries clarified and having decided to willingly take part in the study, each subject signed the Informed Consent Form (ICF) previously approved by the Avant Santé Research Center Research Committee (COFEPRIS 18 CI 19 019 021) along with the study protocol (approval CONBIOÉTICA-19-CEI-010-20160830). Initially, 32 study subjects were selected and randomized; all the subjects of the study completed the procedures.
Single-dose, randomized, open-label, two-treatment, two-sequence, two-period, crossover bioequivalence study of 10 mg lenalidomide capsules manufactured by Eurofarma Laboratórios S.A. versus Revlimid® (10mg lenalidomide immediate release capsule) manufactured by Celgene Europe B.V. and registered in Brazil by Bristol-Myers Squibb Farmacêutica LTDA in adult male healthy volunteers under fasting conditions.
Study subjects were kept in fasting conditions for 10 hours prior to the dose administration and at least 4 hours afterwards on each period. The study was conducted under fasting conditions as required by Anvisa.
On each period of the study, a single dose (10mg each) of the test or the reference product was administered orally to the volunteers, in a seated position, with 200ml of water at room temperature, under fasting conditions. The washout period was 7 days.
A total number of 19 blood samples were collected of each volunteer in each period in tubes with K2EDTA. Blood samples (4ml each) were collected at timepoints 0.00h (before dose) and after administration at timepoints 0.25, 0.33, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75, 2.00, 2.50, 3.00, 4.00, 6.00, 8.00, 10.00, 12.00, 16.00 and 24.00 hours.
After collection, blood samples were processed in refrigerated centrifuge at 3,000rpm for 10 minutes at 4±2ºC. The plasma obtained from the blood samples was transferred to two different cryogenic tubes (aliquot 1 and aliquot 2) previously identified and stored at a temperature below -50°C.
The validation of the bioanalytical method for quantification of Lenalidomide in human plasma using Lenalidomide-d5 as internal standard and K2EDTA as anticoagulant through extraction in solid phase (Strata-X® 33µm cartridge) and liquid chromatography coupled to mass spectrometry (UPLC-MS/MS) was performed in compliance with the acceptance criteria for selectivity, calibration curve, precision, accuracy, residual effect, matrix effect and stability test in solution and in the biological matrix.
Chromatographic conditions adopted for the validation and quantification of the study subjects’ samples included the use of a Luna omega 1.6µm PS C18 50x2.1mm chromatographic column, at 35±2 ºC. Samples were kept at 5±4 ºC in the sampler. Mobile phase A used was Formic Acid 0.1% and mobile phase B was LC-MS grade Acetonitrile in an 87:13 v/v proportion. The injection volume was 3.0 µL and retention times were 1.07±0.3 min for the analyte and 1.05±0.3 for the internal standard, the running time being 2.10 minutes.
The method proved linear between concentrations of 2.044ng/ml to 1015.506ng/ml according to equation y = a + bx [1/x], where “y” is the response, “x” is the analyte concentration and “1/x” is the selected weight.
The lower limit of quantification (LLOQ) established for the method was 2.044ng/ml and validated quality control samples were 5.148ng/ml, 411.867ng/ml and 792.052ng/ml.
Stability analysis was carried out in plasma in concentrations of 5.148ng/ml and 792.052ng/ml and they complied with the acceptance criteria when the samples were subjected to 6 hours and 18 minutes at room temperature (approximately 25ºC) (short-term stability), for 2 days 8 hours and 38 minutes in autoinjector (5±4ºC) after sample extraction completion (post-processing stability) 4 freeze-and thaw cycles (freezing temperature: -70°C±15°C) and 106 long-term days.
Reference standards: lenalidomide (TRC/Canada) was used as analyte and Lenalidomide-d5 (TRC/Canada) was used as internal standard for the preparation of the primary standard solutions in methanol HPLC grade. Working solutions were prepared using milli-Q water: Methanol (20:80 v/v) as eluent. All solutions were stored at a temperature between 2 and 8ºC.
Compounds were extracted from human plasma samples and quantified through liquid chromatography coupled to mass spectrometry (LC-MS/MS) using the Xevo TQ-S/Acquity UPLC I-Class (Waters) spectrometer, equipped with positive electrospray (ESP+) ionization source, and the analyte and internal standard were detected using MRM with m/z transitions 260.21>149.11 and 265.23>151.13, respectively.
| Analyte | Lenalidomide |
|---|---|
| Internal Standard | Lenalidomide-d5 |
| Biological Matrix | Human Plasma |
| Anticoagulant | EDTA |
| Linearity | 2.044ng/ml to 1015.506ng/ml |
| Curve Equation | y = a + bx [1/x] |
| Lower Limit of Quantification (LLOQ) | 2.044ng/ml |
| Low Quality Control (LQC) | 5.148ng/ml |
| Medium Quality Control (MQC) | 411.867ng/ml |
| High Quality Control (HQC) | 792.052ng/ml |
| Post-processing Stability Time | 2 days 8 hours 38 minutes |
| Freeze/thaw cycles | 4 cycles |
| Short-term stability time | 6 hours 18 minutes |
| Long-term stability time | 106 days |
Software Masslynx version 4.1 was used for calculating sample concentrations in the analytic phase.
Software Phoenix WinNonlin™ version 8.3 and Sistema de Análise Estatística (SAS®) version 9.4 were used to perform the statistical analysis.
The study began with 32 volunteers and ended with 32 adult healthy male volunteers between 18 and 42 years of age and BMI between 18.7 and 26.9kg/m2, who complied with the inclusion and exclusion criteria set forth in the protocol.
Pharmacokinetic parameters Cmax and AUC0-t were established using software Phoenix WinNonlin™ version 8.3 and Sistema de Análise Estatística (SAS®) version 9.4.
Pharmacokinetic parameters are shown in the table below.
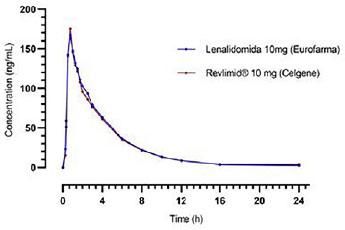
Maximum concentration Cmax obtained for the reference product Revlimid® was 195.309ng/ml in 1.5 hour. For the test product, lenalidomide, Cmax of 186.120ng/ml occurred at 1.1 hour.
Figure 1 Lenalidomide intermediate concentrations along time for each formulation.
Four adverse events were reported during 10mg study: 2 headaches, 1 vomit and 1 dyspepsia. No serious adverse events were reported; 3 events were classified as mild and 1 as moderate.
As to the relation with the study drug, 3 adverse events were classified as probably related and 1 as possibly related.
The study was planned and conducted according to the legal rules and regulations in force, obtaining pharmacokinetic parameters Cmax and AUC0-t which confidence interval values (90%) are within the acceptable limit for the ratio between the geometric means of the test and reference products (80-125%), according to the national legislation.
The planned number of 32 adult healthy male subjects is consistent with studies published by other authors.
The clinical trial was conducted normally, and as mentioned above, there were no serious adverse events. Headache was the most frequent adverse event, contrary to the reports of other authors and the Anvisa panel, who stated neutropenia and thrombocytopenia as the most frequent adverse events.
The 7-day washout period seemed adequate, as all base collection samples of the volunteers of the second period had a concentration below the Lower Quantification Limit (LQL).
As in other published works, the analytical technique selected for the study for lenalidomide quantification of in human plasma samples was LC-MS/MS.
Lenalidomide was quantified in its unaltered form, as required by the Brazilian legislation.
Both reference and test drugs showed a maximum plasma concentration Cmax of 195.13ng/ml and 207.31ng/ml, respectively, consistent with those found in the literature.
In countries with centralized health systems, such as in Brazil (SUS: Sistema Único de Saúde), the importance of generic drugs is based on maintenance of supply and negotiation with pharmaceutical companies. Being so, not only patients benefit from high-quality generic drugs: the savings for the Brazilian health care system from generic drugs is highly expressive. As health care costs continue to rise, it is important to continue to manufacture generic alternatives and make them available to patients, as this may help slow the increase in health care costs which are often passed along to patients.
Taking the results obtained in this study into account, it is concluded that the 10mg lenalidomide capsule test treatment (Eurofarma Laboratórios S/A) and the Revlimid® - 10mg lenalidomide capsule (Celgene Europe B.V.) reference treatment are bioequivalent regarding the absorption rate and extent, when administered under fasting conditions, as the criteria required by the Brazilian regulatory authority has been complied with (CI90% between 80-125%).
According to the statistical results, it can be concluded that both products tested in this study, namely 10mg Revlimid® capsules (reference product) and Lenalidomida Eurofarma (test product), meet the regulatory criteria for bioequivalence. Therefore, based on their biopharmaceutical performance, the test product can be considered interchangeable with the reference product, ensuring their comparability in terms of therapeutic effect and safety.
Based on adverse events and their severities, and clinical examination, electrocardiogram, and laboratorial assays, both products were considered well tolerated by the participants. The found adverse events’ profile was in accordance with the reported in the literature and insert package reference product.
Finally, the use of generic drugs in the clinical practice must be encouraged and be an alternative for public health systems to reduce costs and keeping quality of offered treatment, once bioequivalence trials show interchangeably between generic and reference ones.
| Ratio (test/reference) | Geometric Mean (%) | CI (90%) | Test Power (%) | p-value (sequence) |
|---|---|---|---|---|
| C max (ng/mL) | 95.3 | 84.01 - 108.10 | 89.9 | 0.8631 |
| AUC0-t (ng.h.ml-1) | 101.9 | 98.58 - 105.34 | 100.0 | 0.2633 |
CS Data analysis and interpretation, Manuscript writing, Final approval of the manuscript.
VMR Manuscript writing, Final approval of the manuscript.
LCV Conception and design, Provision of study materials or patient, Final approval of the manuscript.
NCC Collection and assembly of data, Conception and design, Final approval of the manuscript.
MECL Collection and assembly of data, Conception and design, Final approval of the manuscript.
MHB Collection and assembly of data, Conception and design, Final approval of the manuscript.
SK Collection and assembly of data, Conception and design, Final approval of the manuscript.
MP Collection and assembly of data, Conception and design, Final approval of the manuscript.
LB Conception and design, Final approval of the manuscript.
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.
Journal: Brazilian Journal of Oncology
DOI: 10.1055/s-00059887
e-issn: 2526-8732
Publisher: Thieme Revinter Publicações Ltda.
Publisher address: Rua do Matoso 170, Rio de Janeiro, RJ, CEP 20270-135, Brazil
No citations found for this article.
1. Agência Nacional de Vigilância Sanitaria (Anvisa). Guia para provas de biodisponibilidade relativa/bioequivalência de medicamentos. Anvisa, 2006.
2. National Library of Medicine (NIH). Compound Summary - CID 216326: Lenalidomide Information.. NIH/PubChem, .
3. Anderson, KC. Lenalidomide and thalidomide: mechanisms of action-similarities and differences. Semin Hematol [online]. 2005, vol. 42, p. S3-S8.
4. Chen, Y and Borthakur, G. Lenalidomide as a novel treatment of acute myeloid leukemia. Expert Opin Investig Drugs [online]. 2013, vol. 22, p. 389-97.
5. US Food & Drug Administration (FDA). Drug Approval Package. 021880 - Revlimid (Lenalidomide) capsules. FDA, 2005.
6. European Medicines Agency (EMA). EMA/13846/2020 - Revlimid lenalidomide. EMA, 2020.
7. Agência Nacional de Vigilância Sanitaria (Anvisa). Revlimid [Internet]. Anvisa, 2017.
8. Agência Nacional de Vigilância Sanitaria (Anvisa). Lenalidomida: autorizadas novas indicações terapêuticas [Internet]. Anvisa, 2020.
9. Armoiry, X and Aulagner, G and Facon, T. Lenalidomide in the treatment of multiple myeloma: a review. J Clin Pharm Ther [online]. 2008, vol. 33, p. 219-26.
10. Fine, HA and Kim, L and Albert, PS and Duic, JP and Ma, H and Zhang, W. A phase I trial of lenalidomide in patients with recurrent primary central nervous system tumors. Clin Cancer Res [online]. 2007, vol. 13, p. 7101-6.
11. Drugs.com. Lenalidomide pregnancy and breastfeeding warnings [Internet]. Drugs.com, 2022.
12. Agência Nacional de Vigilância Sanitaria (Anvisa). Lista 1 - Forma de administração (formas farmacêuticas de liberação imediata). Anvisa, 2022.
13. Lee, S and Hwang, JG and Park, SY and Lim, HJ and Lee, SW and Seo, MH. Single-dose comparative pharmacokinetics of two formulations of lenalidomide 25mg in healthy subjects: a randomized crossover study. Adv Ther [online]. 2018, vol. 35, p. 210-7.
14. Wang, J and Qi, L and Wang, Z and Chen, G and Liu, C and Liu, X. Bioequivalence study of single-dose lenalidomide capsule vs. Revlimid((R)) capsule in healthy Chinese males. Cancer Chemother Pharmacol [online]. 2018, vol. 82, p. 159-64.
15. US Food & Drug Administration (FDA). Draft guidance on Lenalidomide [Internet]. FDA, 2013.
16. Agência Nacional de Vigilância Sanitaria (Anvisa). Dispõe sobre o controle da substância lenalidomida e de medicamento que a contenha, e dá outras providências. Anvisa, 2022.
17. Agência Nacional de Vigilância Sanitaria (Anvisa). VigiMed - Eventos Adversos [Internet]. Anvisa, 2022.
18. Gay, F and Hayman, SR and Lacy, MQ and Buadi, F and Gertz, MA and Kumar, S. Lenalidomide plus dexamethasone versus thalidomide plus dexamethasone in newly diagnosed multiple myeloma: a comparative analysis of 411 patients. Blood [online]. 2010, vol. 115, p. 1343-50.
19. Ranganathan, P and Gunasekaran, V and Singhvi, I and Ansari, MJ. Development and validation of Lenalidomide in human plasma by LC-MS/MS. Saudi J Biol Sci [online]. 2019, vol. 26, p. 1843-7.
20. Iqbal, M and Wani, TA and Khalil, NY and Darwish, IA. Development and validation of ultra-performance liquid chromatographic method with tandem mass spectrometry for determination of lenalidomide in rabbit and human plasma. Chem Cent J [online]. 2013, vol. 7, p. 7.
21. Hasnain, MS and Rao, S and Singh, MK and Vig, N and Gupta, A and Ansari, A. Development and validation of LC-MS/MS method for the quantitation of lenalidomide in human plasma using Box-Behnken experimental design. Analyst [online]. 2013, vol. 138, p. 1581-8.
22. Agência Nacional de Vigilância Sanitaria (Anvisa). Lista 2 - Analito para Estabelecimento da Biodis-ponibilidade Relativa/Bioequivalência. Anvisa, 2022.
Dados de acesso insuficientes para visualização no mapa.