Ewing sarcoma family of tumors are rare malignancies derived from a common cell of origin.
PNET (primitive neuroectodermal tumor) first called peripheral neuroepithelioma and Askin's tumor of the chest wall, is a more differentiated member of this tumor family. It was first described in 1918 by Stout as a tumor of the ulnar nerve, comprising characteristics of a sarcoma, but composed of small round cells arranged as rosettes.
PNET can present with a wide range of clinical features depending on the affected site, though pain and swelling of the surrounding structures are the more common signs and symptoms.
It is recognized as an aggressive tumor, with an accelerated progression and high relapse rate, requiring an multiprofessional approach in term soft treatment.
PNET occurs more frequently in individuals aged 10 to 20 years, thus being considered as child and adolescent neoplasm.
Disease occurrence in older patients is considered an adverse prognostic factor, although it is not known if this is a consequence of the biological differences or differences in treatment approach. There is some evidence that older patients treated the same way as the younger ones may have comparable survival outcomes
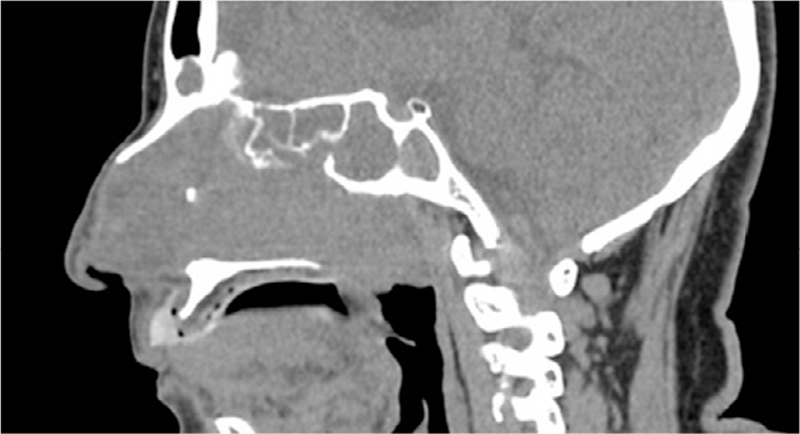
A 69-year-old female presented with complains of swelling and pain in the nasal cavity. She was submitted to Nasal Sinus's CT (Computed Tomography) about 6 months after symptoms onset. Images revealed an expansive mass with soft tissues density occupying completely the maxillary, frontal, ethmoidal and left sphenoidal sinuses, as well as the nasal cavity and fossa, also extending itself to the left side coana and rhinopharynx, diminishing the air column. There was no cleavage plane with the left nasal turbinates, though it was clear that the adjacent osseous areas were remodeled and thinned including the maxillary sinus's posterior wall (
Figure 1 Expansive lesion with soft tissue density and permeation calcifications occupying all paranasal sinuses on the left, besides the fossa and nasal cavity, with extension to the coana and rhinopharynx.
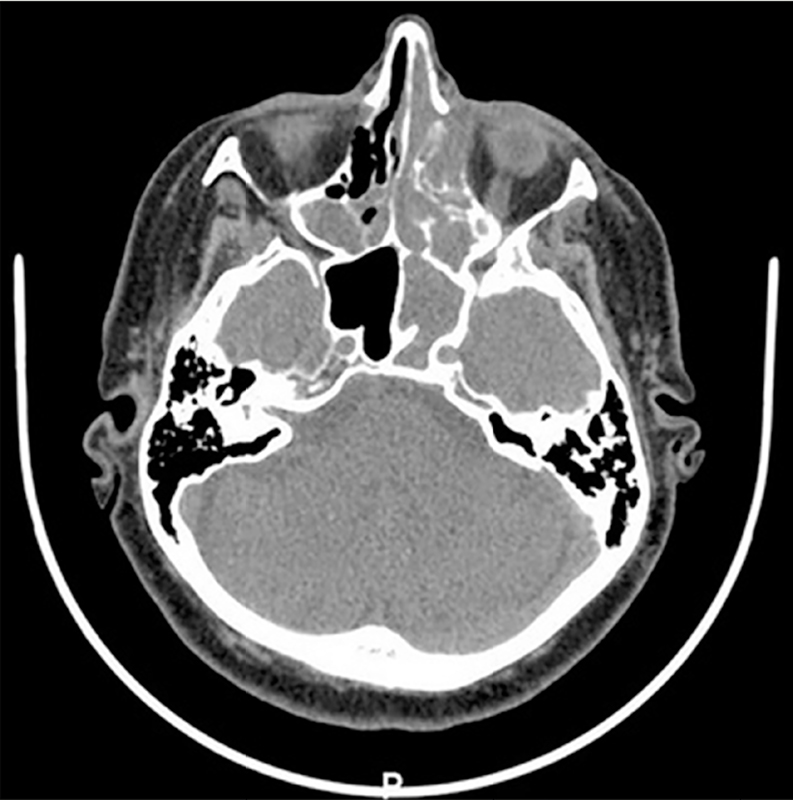
It was also evidenced an infundibular enlargement of the Ostium meatal complex and bulging of the papyracea blade on the left side. CT scan indicated calcification are as associated as well, which brought, at first, the hypothesis of a nasal sinus polyposis (
Figure 2 Determines bone remodeling with adjacent erosion, infundibular widening of the ostiomeatal complex and bulging of the papyracea blade on the left.
Three months later, an endoscopic nasal exploration with biopsy - sinusectomy with Caldwell Luc technic - was performed. The anatomopathological exam revealed a Large Cells Malignant Undifferentiated Neoplasm on the sinus mucosa with positive border, and the immunohistochemistry study demonstrated a primitive neuroectodermal tumor pattern with cell proliferation index of 50-55%.
At the time of diagnosis, the patient had a decent performance status (ECOG 1) and no significant comorbidities, being a candidate for aggressive treatment. Patient's pain was controlled with oral morphine and systemic chemotherapy was initiated. Patient was prescribed doxorubicin plus cyclophosphamide plus vincristine (VAC) on odd cycles, alternating with iphosphamide plus etoposide on even cycles (VAC/IE regimen).
Unfortunately, after being submitted to 2 cycles of chemotherapy, the patient died from a septic shock.
To our knowledge, this is the oldest reported patient reported with PNET in medical literature. PNETs are usually classified as a child or adolescent disease, but as shown, it may occur in older patients as well.
PNET diagnosis is performed through histological analysis,
Most guidelines for PNETs treatment come from data on children and adolescents. Local control provides a survival benefit for non-metastatic disease, but all patients should receive systemic chemotherapy
The VAC/IE regiment has shown superior overall survival for non-metastatic patients and superior progression-free survival for all patients when compared to VAC alone.
Lack of consistent evidence, poor performance, and worse treatment tolerance may account for a less favorable prognosis when compared to the same disease in younger patients. However, there is some evidence that this disease is still chemosensitive in these patients. A recent retrospective study
PNET tumors are typically a children and adolescent disease, but as our report shows, it can also appear in older populations - such as this 69-year-old woman.
Thus, PNET should not be ruled out as a differential diagnosis in older patients based on age alone. Treating such patients with PNETs is a clinical challenge, but aggressive chemotherapy should not be denied to them.
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.
Journal: Brazilian Journal of Oncology
DOI: 10.1055/s-00059887
e-issn: 2526-8732
Publisher: Thieme Revinter Publicações Ltda.
Publisher address: Rua do Matoso 170, Rio de Janeiro, RJ, CEP 20270-135, Brazil
No citations found for this article.
No references with the required fields found.
Dados de acesso insuficientes para visualização no mapa.